Example with custom classification of genotype hits
Natacha Couto
2026-03-12
Source:vignettes/StaphAureusClindamycin.Rmd
StaphAureusClindamycin.RmdAnalysing clindamycin resistance in Staphylococcus aureus
Introduction
This is document summarises the analysis of clindamycin resistance in S. aureus. More specifically, we will look at the distribution of clindamycin resistance in S. aureus isolates from a dataset of clinical samples.
Start by loading the package:
Data preparation
For this example, we have collated genotype-phenotype data from NCBI
and EBI through the ESGEM-AMR Staphylococcus subgroup. Phenotypic data
were collated into a single table (one row per isolate). The genotype
data was generated using AMRFinderPlus. The pre-loaded objects
pheno_CLI_public and afp_CLI_public serve as
the input for AMRgen. First, we will
create a new variant.label column that includes the marker
gene name and its closest accession number, and also indicate whether
the match to the accession is exact or not.
# Create the new marker.label column
afp_CLI_public <- afp_CLI_public %>%
mutate(exact_match = if_else(
`% Coverage of reference sequence` == 100 &
`% Identity to reference sequence` == 100.00,
"", "x_"
)) %>%
mutate(node_hit = paste0(node, "__", exact_match, `Accession of closest sequence`)) %>%
mutate(variant.label = case_when(
`variation type` == "Inactivating mutation detected" ~ paste0(node_hit, ":-"),
!is.na(mutation) ~ paste0(node_hit, ":", mutation),
TRUE ~ node_hit
))Genotype-phenotype analysis
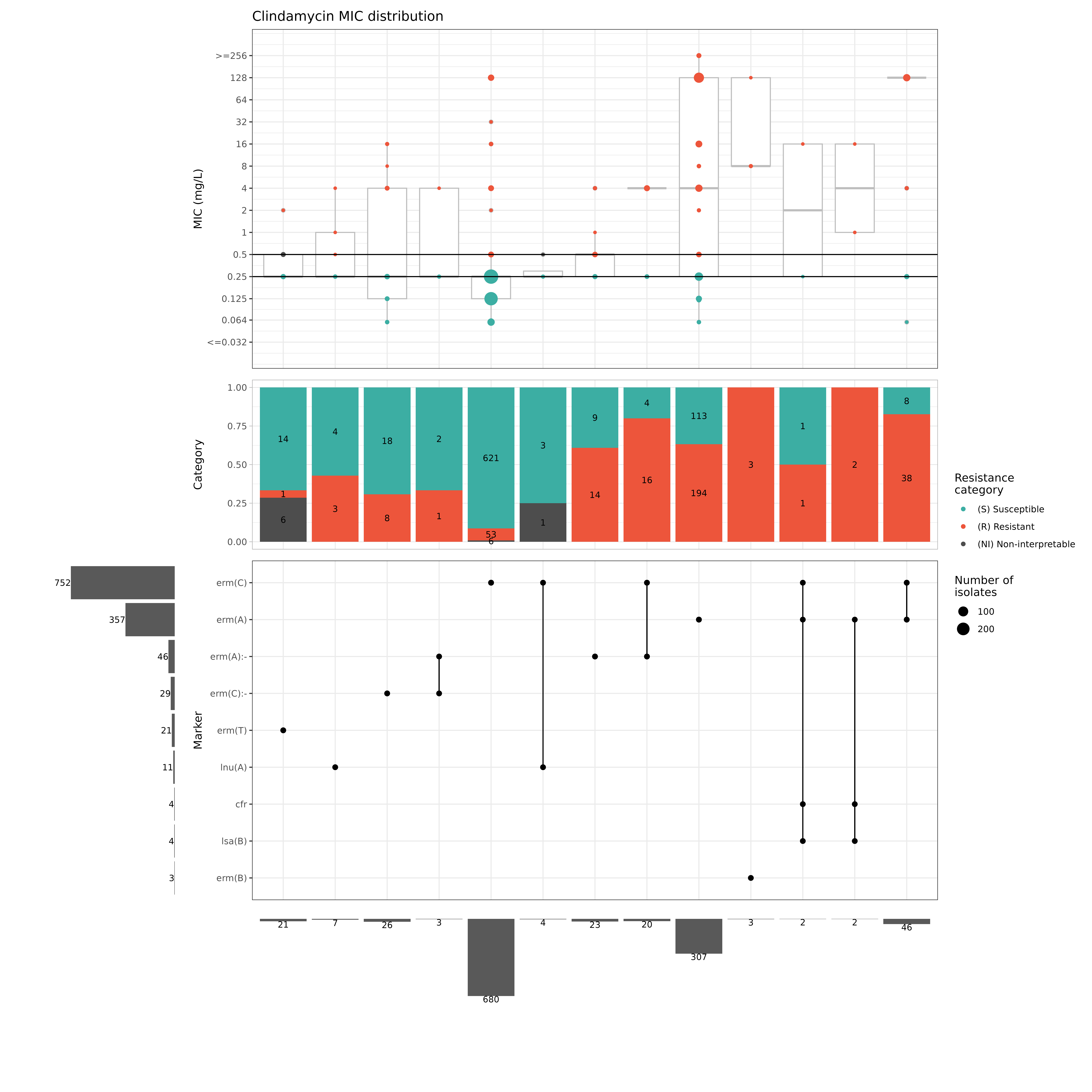
Now let’s create some upset plots, to see how genes, and their allelic variants, relate to clindamycin MICs.
# Visualise with UpSet plot (markers)
cli_mic_upset <- amr_upset(
geno_table = afp_CLI_public,
pheno_table = pheno_CLI_public,
marker_col = "marker.label",
pheno_drug = "Clindamycin",
min_set_size = 2,
assay = "mic",
order = "value",
print_set_size = TRUE,
plot_set_size = TRUE,
print_category_counts = TRUE,
bp_S = 0.25,
bp_R = 0.5
)
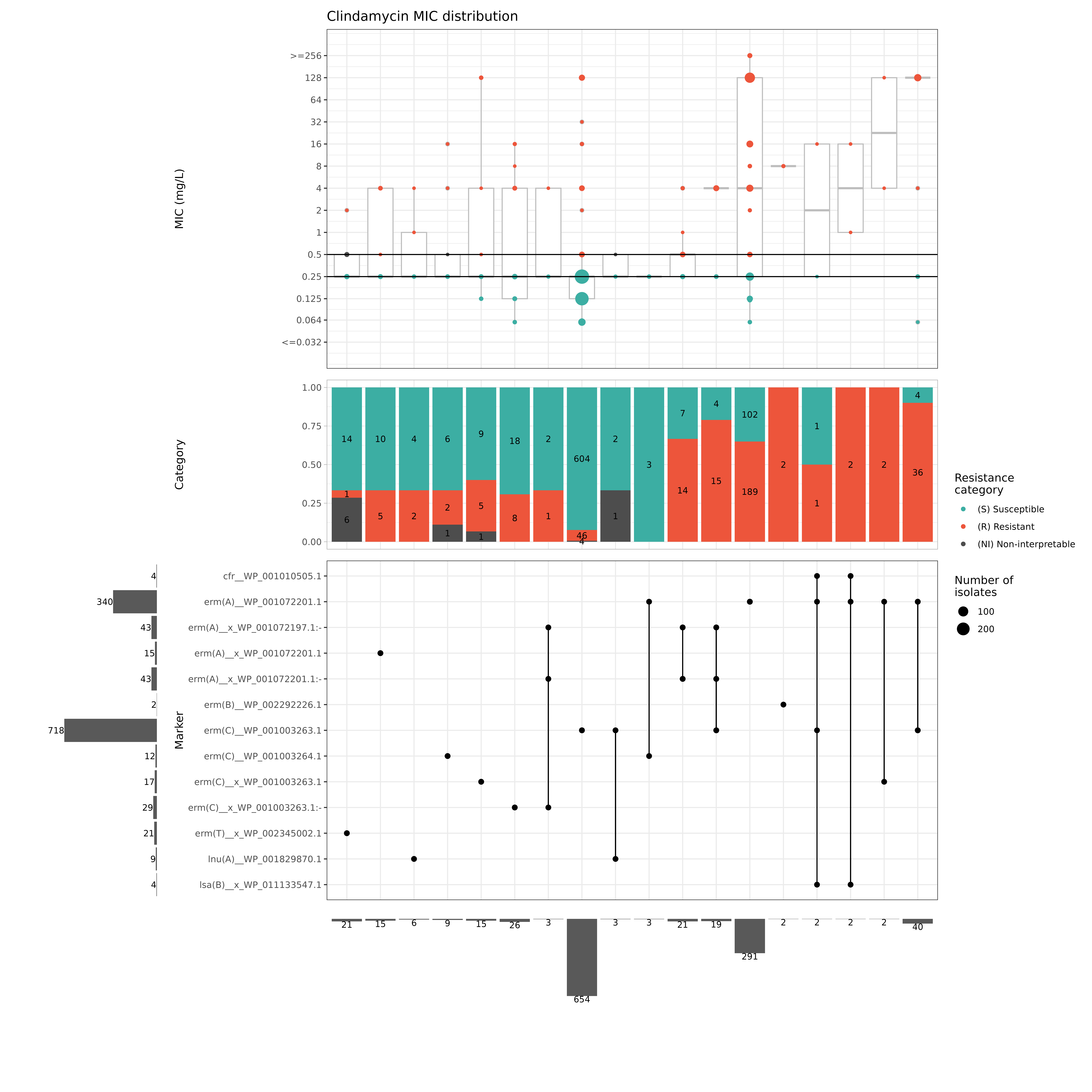
# Visualise with UpSet plot (markers)
# order markers alphabetically so we can more easily see where there are different variants of the same gene
cli_mic_upset_variant <- amr_upset(
geno_table = afp_CLI_public,
pheno_table = pheno_CLI_public,
marker_col = "variant.label",
pheno_drug = "Clindamycin",
min_set_size = 2,
assay = "mic",
order = "value",
marker_order = "alpha", # order markers alphabetically
print_set_size = TRUE,
plot_set_size = TRUE,
print_category_counts = TRUE,
bp_S = 0.25,
bp_R = 0.5
)
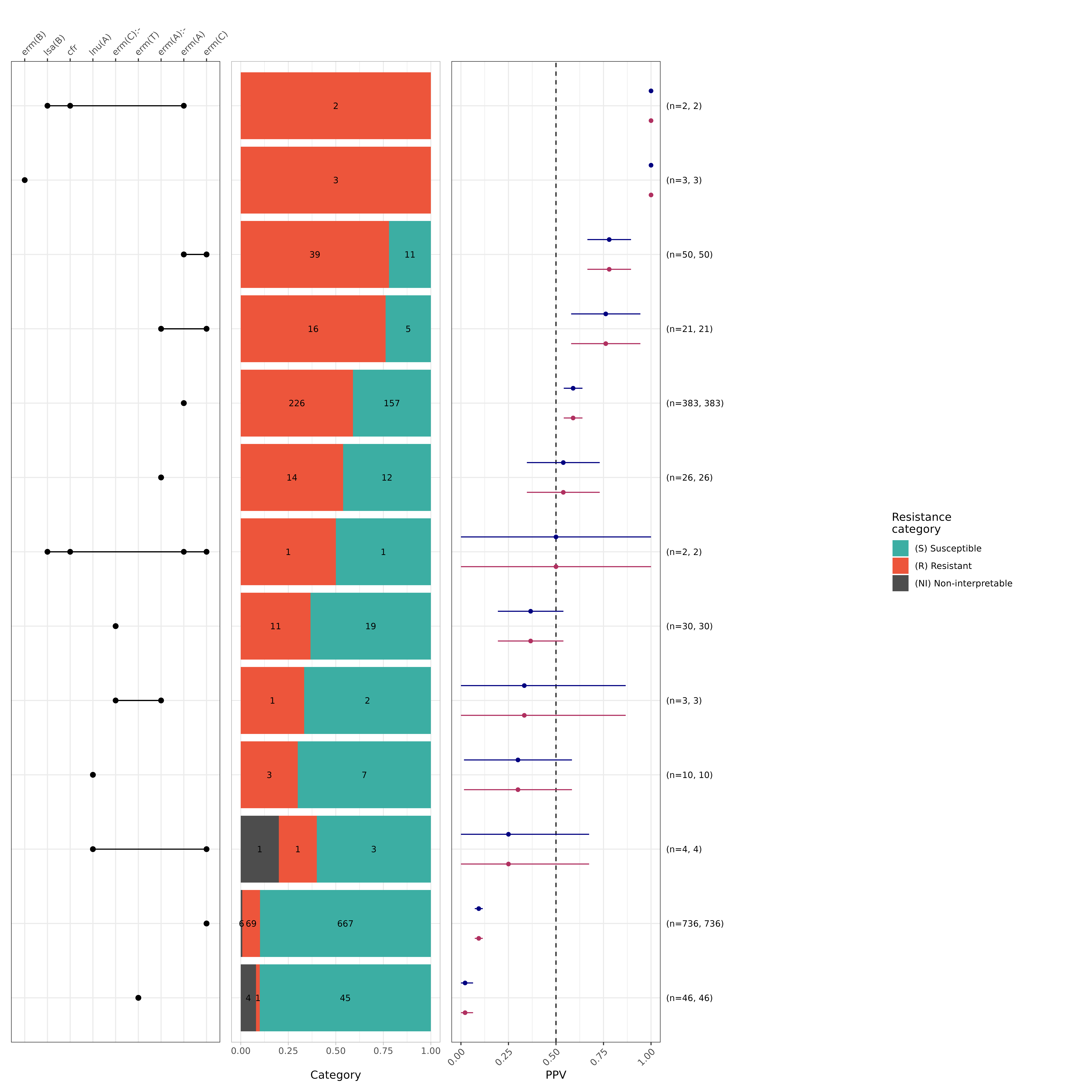
Solo PPV analysis
Next, we will calculate the solo positive predictive value (soloPPV) for each marker and variant. This will allow us to see which markers and variants are most predictive of clindamycin resistance.
# soloPPV analysis for markers
# order markers alphabetically so we can more easily see where there are different variants of the same gene
cli_soloPPV <- solo_ppv(
geno_table = afp_CLI_public,
pheno_table = pheno_CLI_public,
marker_col = "marker.label",
pheno_drug = "Clindamycin",
order_ppv = FALSE
)
# soloPPV analysis for variants
# order markers alphabetically so we can more easily see where there are different variants of the same gene
cli_soloPPV_variant <- solo_ppv(
geno_table = afp_CLI_public,
pheno_table = pheno_CLI_public,
marker_col = "variant.label",
pheno_drug = "Clindamycin",
order_ppv = FALSE
)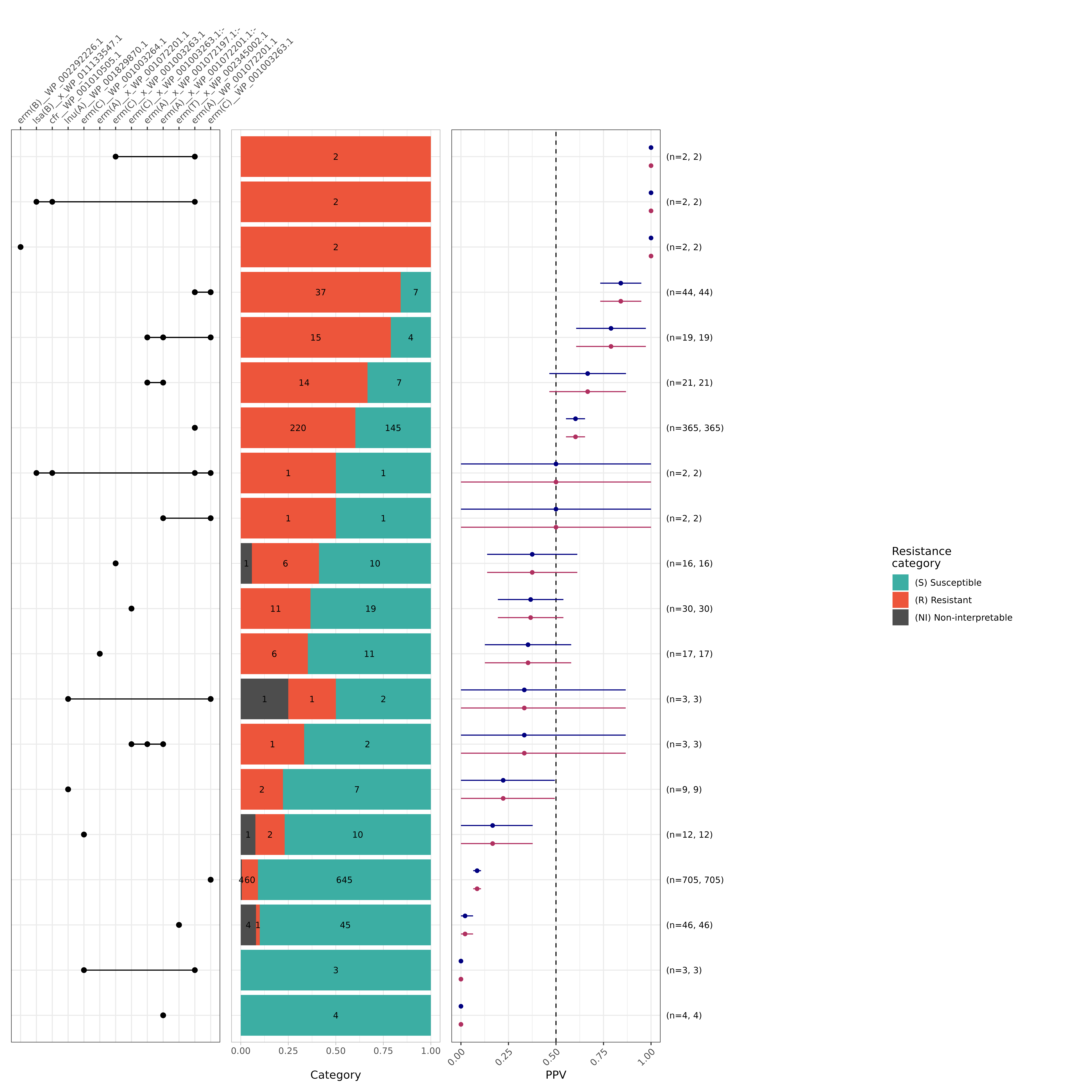
cli_soloPPV_variant$solo_stats
## # A tibble: 38 × 8
## marker category x n ppv se ci.lower ci.upper
## <chr> <chr> <dbl> <int> <dbl> <dbl> <dbl> <dbl>
## 1 erm(A)__WP_001072197.1 R 0 1 0 0 0 0
## 2 erm(A)__WP_001072201.1 R 220 365 0.603 0.0256 0.553 0.653
## 3 erm(A)__x_WP_001072197.… R 0 1 0 0 0 0
## 4 erm(A)__x_WP_001072201.1 R 6 17 0.353 0.116 0.126 0.580
## 5 erm(A)__x_WP_001072201.… R 0 4 0 0 0 0
## 6 erm(B)__WP_001038790.1 R 1 1 1 0 1 1
## 7 erm(B)__WP_002292226.1 R 2 2 1 0 1 1
## 8 erm(C)__WP_001003260.1 R 1 1 1 0 1 1
## 9 erm(C)__WP_001003263.1 R 61 705 0.0865 0.0106 0.0658 0.107
## 10 erm(C)__WP_001003264.1 R 2 12 0.167 0.108 0 0.378
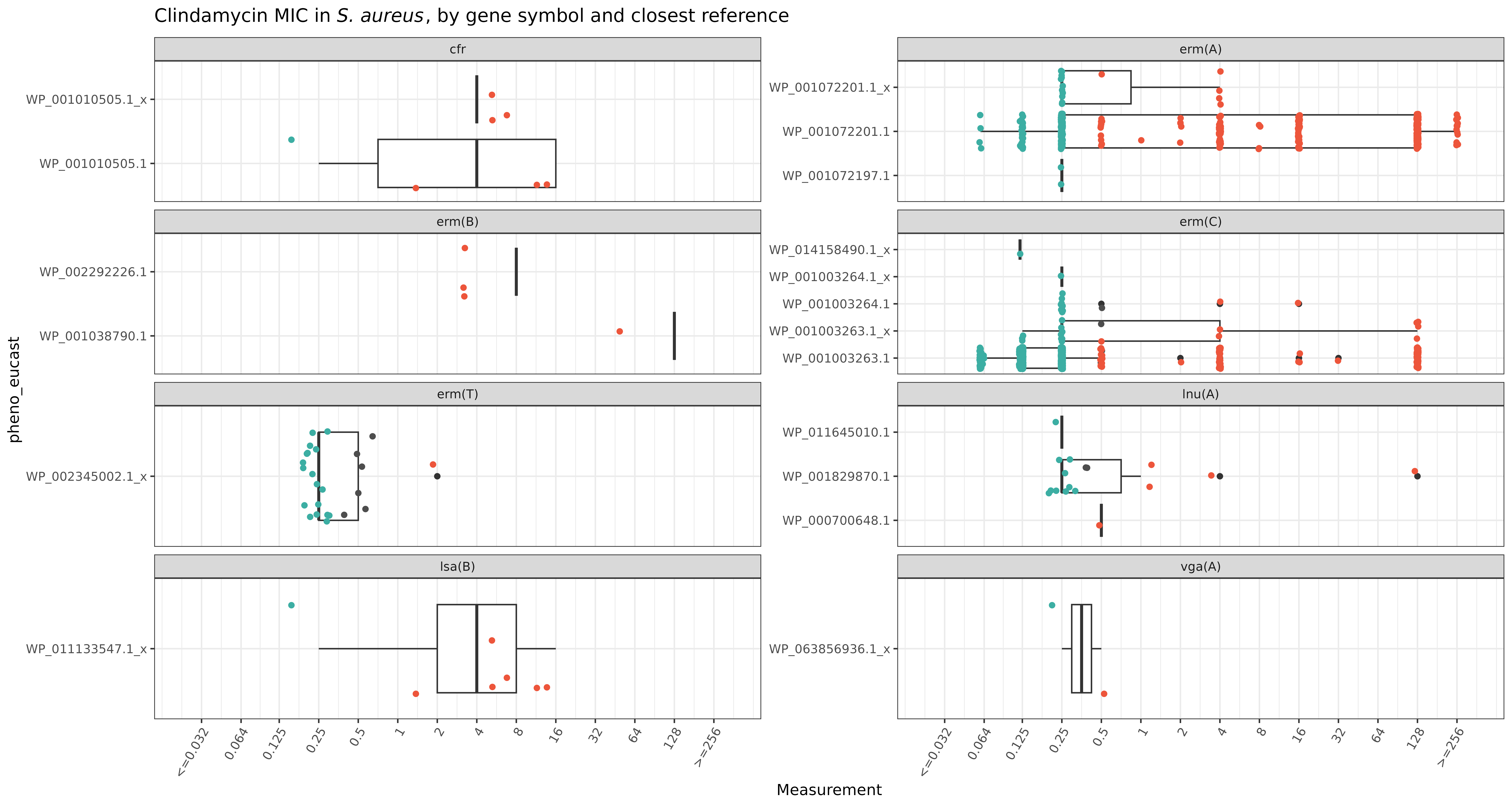
## # ℹ 28 more rowsWe can see from this analysis that the variants of the markers have different associations with clindamycin resistance. For example, the isolates with exact matches to erm(C)_WP001003263.1 or erm(C)_WP001003264.1 are associated with low PPV for resistance (8.5%, n=60/705 and 17%, n=2/12, respectively). This highlights the importance of considering the specific variants of the markers when predicting clindamycin resistance. Note: this discrepancy may be caused by inducible resistance, which is not always captured by a standard AST test. This is because the resistance gene may not be expressed under the conditions of the AST test, but can be induced in the presence of certain antibiotics (e.g., erythromycin). This is a known phenomenon for clindamycin resistance in S. aureus, where the presence of an erm gene can lead to inducible resistance that may not be detected in a standard AST test.
MIC distribution associated with allelic variants
We can use the assay_by_var() function to visualise the
MIC distributions associated with different variants of the same gene.
First we need to join the key genotype fields into the phenotype table,
and then use assay_by_var() to plot MIC assay measures
grouped by variant and faceted into one panel per gene, coloured by
S/I/R phenotype.
cli_geno_pheno <- afp_CLI_public %>%
filter(`variation type` == "Gene presence detected") %>%
mutate(variant_hit = if_else(exact_match == "",
`Accession of closest sequence`,
paste0(`Accession of closest sequence`, "_x")
)) %>%
select(id, marker.label, variant_hit) %>%
left_join(pheno_CLI_public)
## Joining with `by = join_by(id)`
## Warning in left_join(., pheno_CLI_public): Detected an unexpected many-to-many relationship between `x` and `y`.
## ℹ Row 7609 of `x` matches multiple rows in `y`.
## ℹ Row 3161 of `y` matches multiple rows in `x`.
## ℹ If a many-to-many relationship is expected, set `relationship =
## "many-to-many"` to silence this warning.
cli_mic_byhit <- assay_by_var(cli_geno_pheno,
group_by = "variant_hit",
boxplot = T, colour_by = "pheno_eucast"
)$plot +
facet_wrap(~marker.label, scales = "free_y", ncol = 2) +
coord_flip() +
theme(legend.position = "none")
cli_mic_byhit + ggtitle(expression(paste(
"Clindamycin MIC in ",
italic("S. aureus"),
", by gene symbol and closest reference"
)))
Association of specific variants with sequence types
Finally, we can also look at the association of specific marker variants with sequence types, if these data are available. This can be done by creating a binary matrix for the variants and then visualizing the associations with a bubble plot.
# Identify "High-Frequency" STs because we have many STs with only a few samples, which can make the plot cluttered and less informative. By filtering to include only STs with a certain number of samples (e.g., n >= 100), we can focus on the most prevalent STs in the dataset, which are likely to provide more meaningful insights into the associations between specific variants and sequence types.
high_freq_sts <- ST_data_CLI %>%
count(ST) %>%
filter(n >= 100) %>%
pull(ST)
# Merge with ST data
cli_accession_st <- afp_CLI_public %>%
inner_join(ST_data_CLI) %>%
filter(ST %in% high_freq_sts)
# Calculate prevalence of variant in each ST
balloon_data <- cli_accession_st %>%
group_by(ST, variant.label) %>%
summarise(n_samples = n_distinct(id), .groups = "drop") %>%
left_join(ST_data_CLI %>% count(ST, name = "total_st_n"), by = "ST") %>%
mutate(percent_prevalence = (n_samples / total_st_n) * 100) %>%
filter(percent_prevalence >= 10)
# Create the Balloon Plot showing only marker-ST combinations with at least 10% prevalence
ggplot(balloon_data, aes(x = ST, y = variant.label)) +
geom_point(aes(size = n_samples, color = percent_prevalence)) +
scale_color_viridis_c(option = "viridis", direction = -1) +
scale_size_continuous(range = c(1, 10)) +
theme_minimal() +
theme(
axis.text.x = element_text(size = 16, angle = 45, hjust = 1),
axis.text.y = element_text(size = 16),
legend.title = element_text(size = 16),
legend.text = element_text(size = 14)
) +
labs(
title = "Clindamycin Markers by Major Sequence Types",
subtitle = "Analysis of STs with n >= 100",
x = "Sequence Type (ST)",
y = "Marker variant",
size = "Sample Count",
color = "Prevalence (%)"
)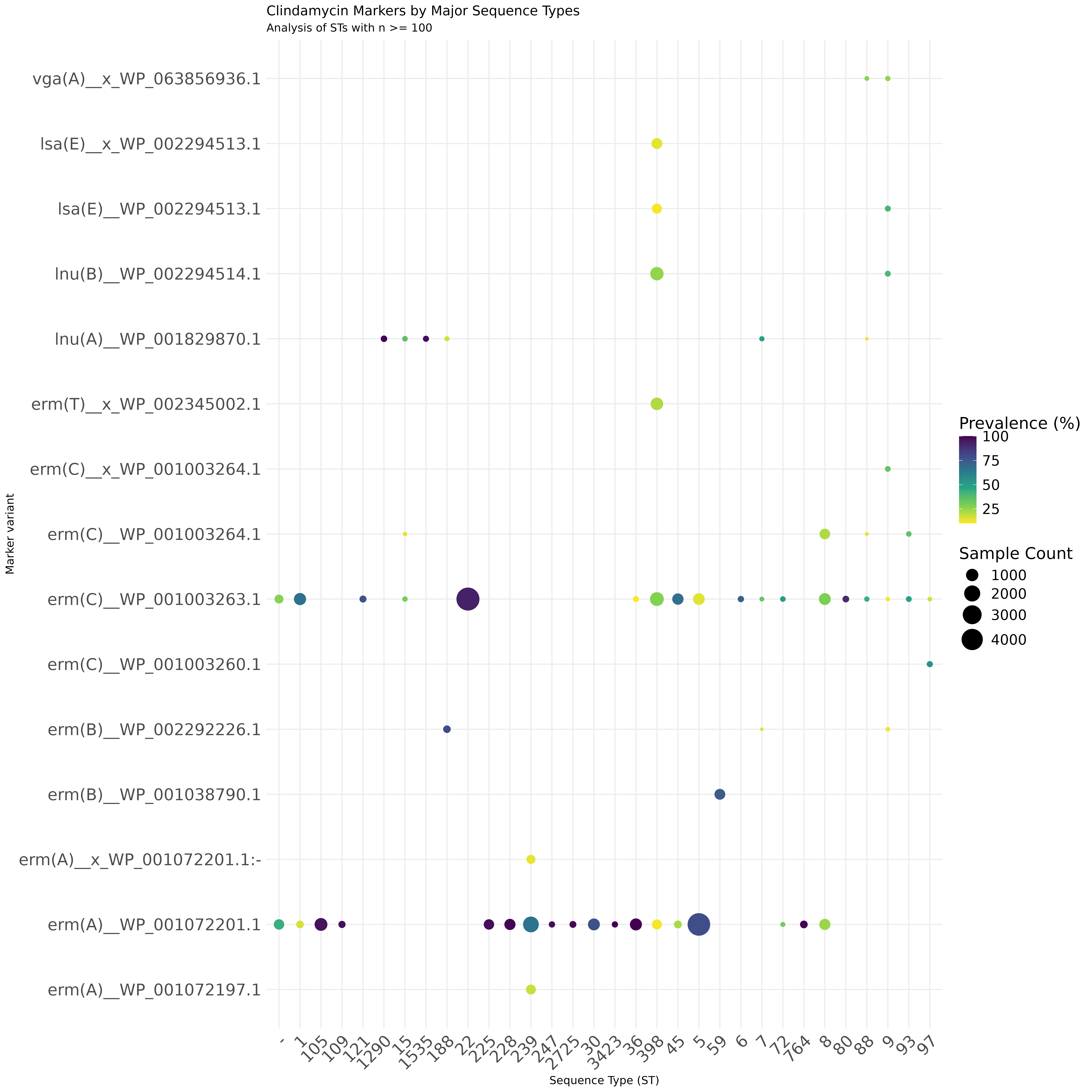
We can see from this last figure, that while erm(C)_WP0012364.1 is quite rare, erm(C)_WP0012363.1 is prevalent in certain STs, such as ST22. This suggests that there may be a clonal association of this variant with this ST, indicating resistance in this clone might be inducible and therefore more difficult to identify using standard AST.